Gene Functional Relationships
Insertional mutagenesis
Insertional mutagenesis is an essential tool to facilitate gene function analysis on a genome-wide scale.
E.g. in maize, the model we focus on, to date only a few hundred of the predicted ~44.000 maize gene models have been functionally characterized. To close this knowledge gap, sequence-indexed reverse genetics collections of loss-of-function mutants for virtually all genes of each genome are required.
While protocols for mutagenesis and flanking sequence tag generation are available for many species and insertion strategies, no tools are accessible for the routine mapping of flanking sequences to genome positions, linking gene annotation to the appropriate seed stocks.
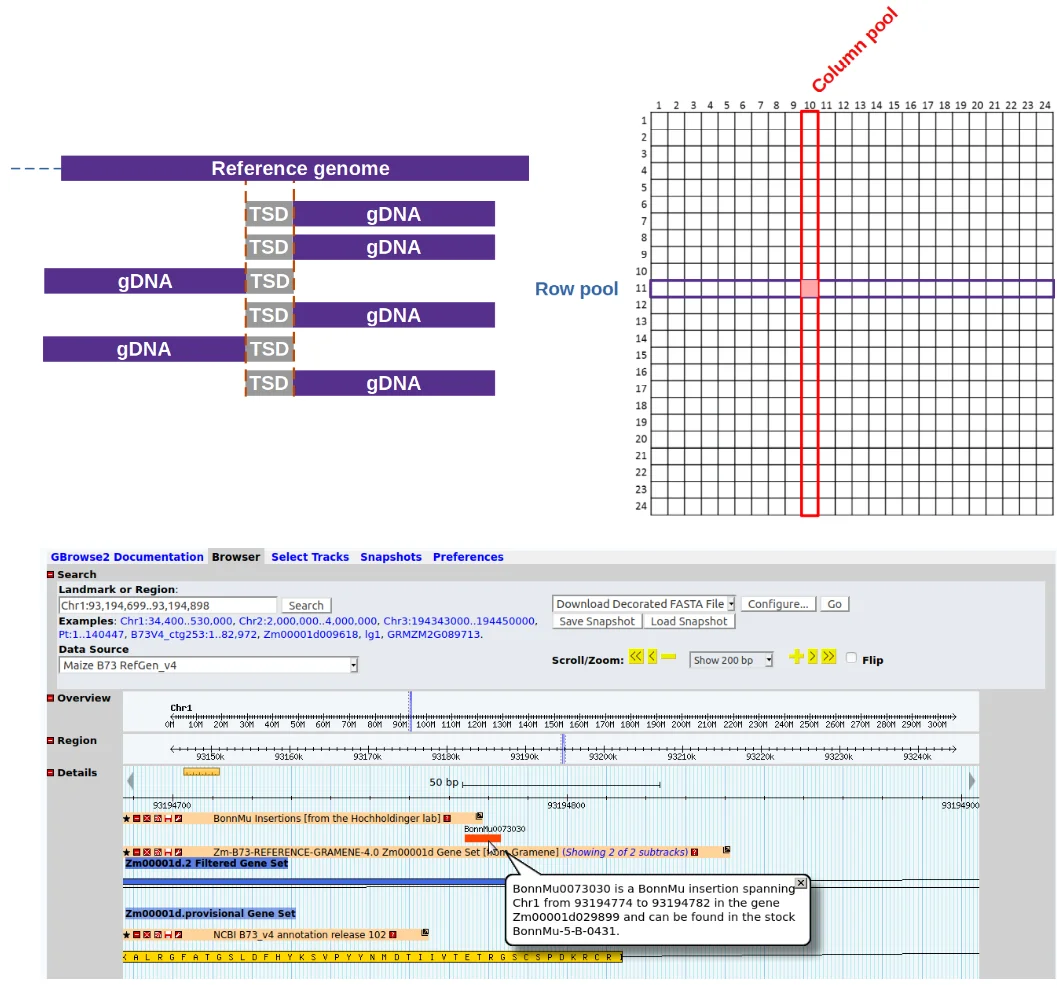
To make loss-of-function mutants for most maize genes more easily available to European researchers, our group recently was part of an effort that introduced a novel Europe based Mutator (Mu) transposon insertional library which was published in Plant Physiology (DOI: https://doi.org/10.1104/pp.20.0047822).
As part of this effort and to accelerate the identification, annotation & analysis of Mu insertions we wrote a bioinformatics software designated MuWU, which has since been expanded to be used as a general tool for downstream analysis & annotation of targeted sequencing of transposon insertion sites.
MuWU - Mutant-seq library analysis and annotation
Automated workflow for the identification and annotation of transposable element insertion sites originally developed for the BonnMu resource and Mutator transposons in particular.
MuWU serves as a fast tool for processing of sequencing reads from an insertional mutagenesis and is able to detect any kind of TE insertion event as long as:
- target site duplications (TSDs) are created by its integration
- the TSD length is known and
- the library is enriched for the areas of insertion/their immediate flanking sequences (e.g. Mutant-seq).
MuWU allows for the reliable identification of insertion tagged genes, taking advantage of a multidimensional pooling design and a TE motif of interest to differentiate germinal from somatic (non-heritable) insertions/mutations.
Find the MuWu Publication in Oxford Bioinformatics here:
Find the MuWu software on GitHub:
Candidate gene prediction for stress adaptation exploring Differential Gene Expression and Gene family expansion
In this project, we predict candidate genes for stress adaptation by integrating differential gene expression under stress and gene family expansion in stress-adapted species. For this purpose, we analyze and compare RNA-Seq datasets and genome sequences of related plant species with different stress tolerance. We use phylogenetic analyses to validate the candidate genes in silico and phenotyping of mutant plant lines to validate the candidate genes in vivo.

We develop A2TEA, a bioinformatic tool which automatically prioritizes these candidate genes for stress adaptation from differentially expressed genes. The integrated A2TEA.Webapp allows the user to interactively explore their RNA-Seq data and view phylogenetic trees of expanded gene families alongside the differential expression.
Find the A2TEA publication and the A2TEA Software on GitHub
We identified interesting candidate genes for drought adaptation in Brassicaceae using A2TEA: